Syndroomia
ja
geenejä
Suomen otolaryngologiyhdistyksen syyskoulutuspäivillä 2009 Kuopiossa saatiin perustietoa genetiikasta Suomen lääketieteellisen genetiikan yhdistyksen asiantuntijoilta. Esitykset olivat erinomaisia. Jäin kuitenkin kaipaamaan selvitystä omaa erikoisalaamme koskevista perinnöllisistä sairauksista ja oireyhtymistä. Yritän tällä kirjoitelmalla paikata puutetta. Kirjoitelma on lyhyt lista eräistä perinnöllisistä oireyhtymistä. Oireyhtymän tyypilliset piirteet käydään lyhyesti läpi ja kerrotaan, mitä sen periytyvyydestä tiedetään. Täydelliseen luetteloon en pyri. Tietolähteenä on käytetty internetiä.
Ymmärrys genetiikan merkityksestä sairauksien taustatekijänä on jatkuvasti kasvamassa. Perinnöllisyyden merkitys ARHI:n (age related hearing impairment) taustalla ja vaikkapa lasten korvatulehdusten taustatekijänä on selvitystyön alla. Vielä ei ole keinoja perimäämme muuttaa mutta jo nyt esimerkiksi farmakogenetiikka vaikuttaa käytännön työssä esimerkiksi varfariinilääkitystä aloitettaessa. Voi olla, että pian testaamme potilaan mahdollisen 1555A>G geenivian, ennen kuin aloitamme aminoglykosidihoidon tai intratympanaalisen gentamysiinihoidon. Tieto voi auttaa vaikean kuulovaurion estämisessä. Tieto potilaan CYP2D6 geenistä voisi kertoa kuinka hän metaboloi kodeiinia tai tramadolia. Tieto auttaisi valitsemaan oikean kipulääkityksen leikkauspotilaalle. Geenitesti kertoo myös alttiudestamme saada laskimoveritulppa esim. leikkaustoimenpiteen komplikaationa ja auttaa hepariinisuojalääkitystä tarvitsevien potilaiden valinnassa.Jos tietäisimme potilaan periytyvästä keloiditaipumuksesta saattaisimme käyttää tietoa hyväksemme hoitovaihtoehtoja punnitsessamme.
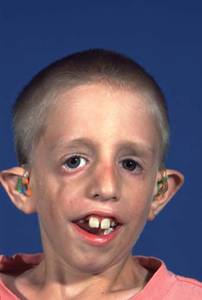
Treacher Collins
Oireyhtymän kuvasi vuonna 1900 brittiläinen oftalmologi ja kirurgi Edward Treacher Collins. Treacher Collins oireyhtymä periytyy autosomaalisti dominantisti. Kraniofakiaaliset poikkeavuudet ovat sille tyypillisiä. Sen yleisyys on 1 / 10 000.
Oireyhtymän syynä on mutaatio TCOF1 geenissä, joka sijaitsee lokuksessa 5q32-q33.1. Geeni koodaa treacle-proteiinia, jonka katsotaan vaikuttavan sikiönkehityksen aikana erityisesti pään ja kasvojen alueella.
Syndrooman oireet vaihtelevat yksilöstä toiseen ja saattavat olla melkein huomaamattomatkin. Keksikasvojen ja alaleuan pieni koko saavat nenän näyttämään kookkaalta ja terävältä. Leuan pieni koko voi työntää vastasyntyneen kieltä taaksepäin ja aiheuttaa hengitysvaikeutta.
Waardenburg
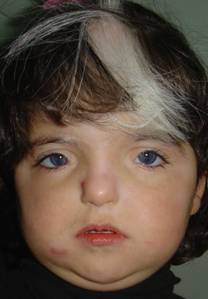
Oireyhtymän kuvasi vuonna 1951 hollantilainen silmälääkäri Petrus Johannes Waardenburg. Oireyhtymän yleisyys on 1 / 42 000 mutta kuurojen koulun oppilailla sen yleisyydeksi on arvioitu 1 / 30. Waardenburgin oireyhtymä periytyy autosomaalisti dominantisti. Oireyhtymä on jaoteltu neljään alatyyppiin ja pieni osa tyyppi II ja IV:sta näyttää periytyvän autosomaalisti resessiivisesti.
Tyyppi I:ssä ja III:ssa mutaatio on PAX3 geenissä, lokus 2q35. Myös muiden alatyyppien geenivirhe tunnetaan.
Alport
Oireyhtymän kuvasi brittiläinen sisätautilääkäri Cecil A. Alport vuonna 1927. Sen yleisyys on 1 / 5000. Syynä oireyhtymään on mutaatio basementmembraanin tyyppi IV kollageenia koodaavissa geeneissä COL4A3, COL4A4 tai COL4A5. Viimeksi mainittu sijaitsee X-kromosomissa ja siksi poikien oireet ovat pahemmat kuin tyttöjen oireet. Miehillä oireyhtymä johtaa munuaisen vajaatoimintaan mutta naisilla glomerulonefriitin aste on vähäisempi ja aiheuttaa usein hematuriaa mutta ei munuaisen tuhoutumista. Koska geeni sijaitsee X-kromosomissa, se ei luonnollisestikaan periydy isältä poikalapsille.
COL4A3 ja COL4A4 sijaitsevat kromosomissa 2 ja periytyvät autosomaalisti resessiivisesti.
Kuulovika esiintyy tavallisesti 2000 – 8000 Hz alueella ja etenee hitaasti. Se harvoin realisoituu ennen kolmattakymmenettä ikävuotta.
Usher
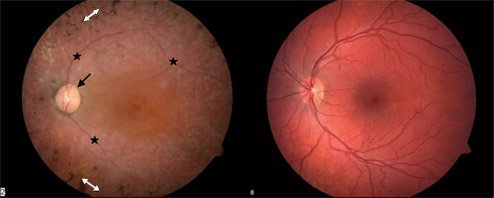
Vasemmalla retinitis pigmentosa (Usher) ja oikealla terve simänpohja
Oireyhtymän kuvasi brittiläinen silmälääkäri Charles Usher vuonna 1914. Oireyhtymä on tärkein kuurosokeuden aiheuttaja ja sen yleisyys on noin 1 / 20 000. Syndrooman taustalla on mutaatio yhdessä kymmenestä eri geenistä. Periytyminen on autosomaalinen resessiivinen. Näkökyvyn menetys johtuu retinitis pigmentosasta. Näkösolujen degeneraatio alkaa yleensä laidoilta ja etenee kohti makulaa. Näköongelma alkaa usein hämäräsokeutena ja etenee sitten tunnelinäöksi ja mahdollisesti sokeuteen asti.
Syndroomalla on kolme kliinistä alatyyppiä:
Tyyppi I usherpotilaat syntyvät vaikeasti kuulovikaisina ja alkavat menettää näkökykyään jo alle kymmenenvuotiaina. Heillä esiintyy tasapainovaikeuksia ja kävelemisen oppiminen viivästyy. Maailmanlaajuisesti tämän tyypin yleisyydeksi arvioidaan 3-6 / 100 000.
Usher tyyppi I:n taustalla voi olla mutaatio missä tahansa geeneistä: CDH23, MYO7A, PCDH15, USH1C tai USH1G.
Tyyppi II usherpotilaiden kuulo on syntyessä erittäin huono. Harvoin he kuitenkaan ovat kuuroja. He alkavat menettää näköään vasta toisella vuosikymmenellä ja saattavat säilyttää osan näöstään keski-ikään asti. Näillä potilailla ei ole tavallisesti tasapainovaikeutta. Taustalla on mutaatio jossakin geeneistä: USH2A, GPR98 and DFNB31. Usher tyyppi II on vaikeammin diagnostisoitavissa kuin tyyppi I ja sen esiintymistä arvioidaan yleisemmäksi.
Tyyppi III usherpotilaiden kuulo ja näkö heikkenevät vähitellen. Noin puolella potilaista esiintyy tasapainovaikeuksia. Oireyhtymässä on eroja. Osa säilyttää lukemiseen riittävän näön myöhäiseen keski-ikään asti. Tyyppi III:n taustalla on CLRN1 geeni, joka koodaa klariini 1 proteiinia. Sillä on merkitystä sisäkorvan ja retinan toiminnalle. Tyyppi III on tavallisempi Suomessa kuin missään muualla maailmassa.
Osler – Weber – Rendu (Hereditaarinen hemorrhaginen teleangiektasia, HHT)
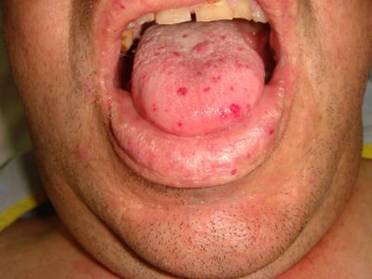
Oireyhtymää luonnehtivat ihon ja limakalvorajojen teleangiektasiat, sisäelinten Av-malformaatiot ja toistuvat nenäverenvuodot.
Diagnoosiin riittävää kolmen neljästä kriteeristä toteutuminen:
Periytyminen on autosomaalinen dominantti. Sen yleisyys on valkoisella rodulla 1 / 5000 mutta mustilla se on äärettömän harvinainen.
HHT:sta erotellaan neljä alatyyppiä. Yhteen niistä liittyy juveliini suolistopolypoosi. HHT:n taustalla arvioidaan olevan geenivirheen aiheuttama angiogeneesin häiriö, joka liittyy sidekudoksen ohjaamiseen ja TGF-ß toimintahäiriöön.
Spesifistä hoitoa ei ole. Suolistovuodot johtavat usein potilaan anemisoitumiseen. Estrogeenilääkityksestä saattaa olla näissä apua. Sisäelinten AV-malformaatioiden hoidossa saatetaan tarvita kirurgiaa tai endovaskulaarisia toimenpiteitä (coilaus). Nenäverenvuodot ovat ongelma johon yksiselitteistä ja selkeää hoitoa ei ole. Lähinnä käytetään paikallisia kauterisaatioita.
Jervell – Lange – Nielsen
Oireyhtymän periytyminen on autosomaalinen resessiivinen. Sen yleisyys on vain 5 / 1 000 000. Se on syynä alle kymmenesosaan pitkän QT-ajan oireyhtymistä.
Oireyhtymän aiheuttaa mutaatio geeneissä KCNE1 tai KCNQ1 (90 %). Nämä koodaavat solujen kaliumpumpun toimintaan liittyviä proteiineja. Kaliumpumppu on tärkeä myös sisäkorvan toiminnan kannalta.
Hoitamattomista potilaista puolen arvioidaan kuolevan rytmihäiriöön ennen viidentoista vuoden ikää. Pitkä QT-aika on riski lääkkeiden käytön yhteydessä. Korvalääkäriä koskettavat makrolidiantibiootit, fluorokinolonit ja ”vanhat” antihistamiinit.
Hoitona on tavallisimmin betasalpaajat ja joskus tarvittaessa toimiva sydämentahdistin.
Pendred
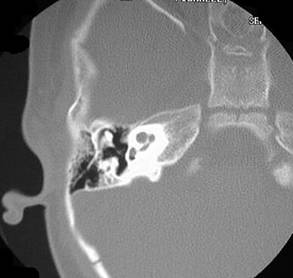
Mondinin dysplasia, koklean kierteet ovat vajaat. Tyvi-
kierre on tallella. Pyöreä ja soikea ikkuna voivat puuttua.
Kalvoinen labyrintti saattaa puuttua tai olla alikehittynyt.
Koklean Reissnerin kalvo on pudonnut alas ja Cortin eli-
messä on leesioita. Stria vaskularis ja spiraaliliga-
mentti ovat normaalit.
Oireyhtymän kuvasi englantilainen lääkäri Vaughan Pendred vuonna 1896. Oireyhtymä on syynä noin 7,5 %:iin synnynnäisistä kuulovioista. Periytyminen on autosomaalinen resessiivinen. Oirekuvaan liittyy synnynnäinen molemminpuolinen sensorineuraalinen kuulovika, joka progredioi. Tämä johtaa puheen kehityksen ongelmiin.75 %:lla potilaista on struuma ja osalla kilpirauhasen vajaatoiminta. Näillä potilailla pään trauma saattaa aiheuttaa äkillisen kuulonlaskun pahenemisen.
Pendredin oireyhtymä liitetään PDS-geenin mutaatioon. Geeni koodaa pendrin-proteiinia. Sitä löytyy kokleasta, kilpirauhasesta ja munuaisesta. Se säätelee bikarbonaatin eritystä mutta oireyhtymään ei kuulu munuaisen toimintahäiriöitä. Saman geenin mutaatio aiheuttaa myös laajan vestibulaarisen aquaduktin syndrooman. Pendred potilailla löytyy TT:ssa usein mondinin dysplasia.
HAE (Hereditaarinen angioödeema, Quincken ödeema)
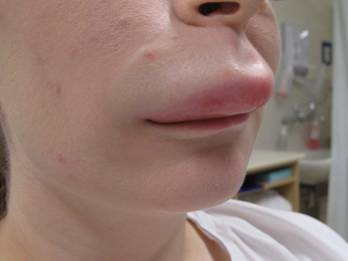
Oireyhtymän kuvasi saksalainen sisätautilääkäri, kirurgi Heinrich Quincke vuonna 1882. Sir William Osler totesi oireyhtymällä olevan perinnöllisiä piirteitä vuonna 1888.
HAE aiheuttaa dermiksen, ihonalaisen kudoksen, limakalvon ja submukosan nopeasti (minuuteista tunteihin) ilmaantuvan turvotuksen, joka voi johtaa potilaan hengenvaaraan ahtauttaessaan hengitysteitä. Turvotukseen voi liittyä kipua tai kutinaa. Turvotus ilmaantuu usein pienen trauman jälkeen. Tällainen voi olla vaikkapa hammastoimenpide. Tavallisesti turvotus häviää 2 – 5 vuorokaudessa. Se ei liity allergiaan tai lääkkeisiin (ACE-estäjät). Adrenaliini, kortisoni tai antihistamiini eivät laukaise kudosturvotusta. Potilailla voi esiintyä myös vatsakipukohtauksia joihin liittyy oksennuksia, ripulia ja heikotusta. Nämä kohtaukset voivat johtaa turhiin kirurgisiin toimenpiteisiin.
HAE:sta tunnetaan kolme eri alatyyppiä:
Tyyppi I:ssa mutaatio on SERPING1-geenissä, joka johtaa C1-inhibiittorin puutokseen. Tämä on yleisin HAE:n muoto ja edustaa 85 %:a HAE:sta.
Tyyppi II:ssa C1- inhibiittorin määrä on normaali mutta toiminta alentunut.
Tyyppi III:ssa mutaatio on hyytymisketjun faktori XII:n toimintaa säätelevässä F12-geenissä. Periytyy X-kromosomin mukana dominantisti. Raskaus tai E-pilleri voivat laukaista oireyhtymän.
HAE tyyppi I ja II periytyvät autosomaalisti dominantisti. C1-inhibiittorin puutos johtaa tavallisesti komplementti 2:n ja 4:n tason madaltumiseen. Bradykiniinilla on tärkeä osa HAE:ssa. Se on vasodilataattori ja lisää suonten permeabiliteettia. Erityisesti kasvojen alueella subkutiksen sidekudos on löyhää ja sallii nopean ja voimakkaan kudosturvotuksen. Bradykiniinin estäminen vähentää HAE-oireita.
Akuutin kohtauksen hoitoon voidaan käyttää C1-INH konsentraattia tai tuoretta jääplasmaa.
Profylaksiaan käytetään Danazolia. Se kuuluu androgeenien ryhmään ja lisää maksan C1-INH tuottoa. Lääkitykseen liittyy usein sivuvaikutuksia. Se ei sovi lapsille eikä raskauden aikana käytettäväksi.
Fibrinolyysin estäjää, traneksaamihappoa, käytetään myös pitkäaikaisprofylaksiaan vaikka sen teho on vaatimaton.
Runsas tunti ennen kirurgisia toimenpiteitä voidaan antaa C1-INH konsentraatti suojalääkitykseksi.
Uutena lääkkeenä on tulossa ekallantidi, joka on jo saanut FDA:n hyväksynnän. Se on kallikreiinia estävä peptidivalmiste. Icatibant on selektiivinen bradykiniinireseptorin salpaaja ja myös tulossa markkinoille.
Di George

Oireyhtymän kuvasi yhdysvaltalainen pediatri Angelo Di George vuonna 1968. Sen yleisyys on 1 / 4000. Oireyhtymän nykyinen nimi 22q11.2 deleetio kertoo, että ongelmana on kromosomin 22 pienen osan puuttuminen. Alla olevassa taulukossa on lueteltuna oireyhtymän tavallisimmat löydökset. CATCH22-nimi tulee tavallisimpien löydösten muistilistasta.
Yksilöllinen vaihtelu on tässäkin oireyhtymässä runsasta. Tavallisimpia ongelmia ovat synnynnäinen sydänvika, suulaen halkio tai vajaatoiminta, oppimisen vaikeudet, infektio-ongelmat ja kasvojen pienet poikkeavuudet. Synnynnäinen sydänvika tai hypokalsemiaan liittyvät vastasyntyneen kouristelut usein johtavat oikeille jäljille jo heti syntymän jälkeen. Mikrodeleetioilla 22q11.2 alueella on myös yhteys huomattavaan skitsofrenian riskin kasvuun. Neljännekselle skitsofrenia kehittyykin ja vielä suurempi osa potilaista kokee psykoosijakson.
Oireyhtymä voi periytyä vaikka yli 90 %:ssa kyse on uudesta de novo deleetiosta.
Crouzon
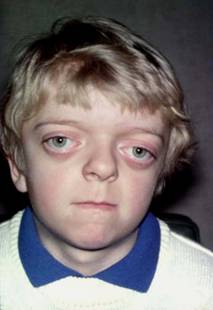
Oireyhtymä on nimetty ranskalaisen lääkärin Octave Crouzonin mukaan. Hän kuvasi oireyhtymän vuonna 1912. Oireyhtymän yleisyys on 1 / 25 000.
Ensisijaisesti oireyhtymässä häiriytyy ensimmäisen kiduskaaren kehitys ja sen myötä keskikasvojen kehitys. Oireyhtymässä kasvojen ja kallon luut liittyvät toisiinsa liian varhaisessa sikiönkehityksen vaiheessa ja tämä estää niiden normaalin laajenemisen. Jo vastasyntyneellä lapsella voidaan poikkeava ulkonäkö todeta. Tässä tyypillisiä löydöksiä:
Kraniofakiaalisella kirurgialla on paljon tarjottavanaan korjausleikkauksissa. Joskus oireyhtymään liittyy sisäkorvaperäinen kuulonlasku ja Menieren taudin oireilu.
Apert

Ranskalainen lääkäri Eugène Apert kuvasi oireyhtymän vuonna 1906. Oireyhtymä voi periytyä autosomaalisti dominantisti mutta tavallisimmin kyse on sporadisista uusista tapauksista, mutaatiosta. Oireyhtymän yleisyys on 1 / 200 000. Oireyhtymän taustalla on fibroblastien kasvutekijään vaikuttavan geenin vaurio kromosomissa 10. Acrocephalosyndactylia kuvaa oireyhtymään kuuluvia löydöksiä. Sikiökehityksen aikana solujen apoptoosi saa alkuvaiheen yhteen kasvaneet sormet erkaantumaan toisistaan. Apertin oireyhtymässä tämä prosessi häiriintyy. Kasvojen ja kallon kehityshäiriö on seurausta sikiökautisesta luuston liian aikaisesta synostoosista eli luiden liittymisestä toisiinsa. Se johtaa tasaiseen takaraivoon ja eteen työntyvään korkeaan otsaan. Kallonsisäisen paineen kasvu voi johtaa aivoston vaurioon ja henkiseen jälkeenjäämiseen. Korvalehdet ovat matalalla ja suulaki korkea, kapea. Keskikasvojen jäädessä pieneksi voi syntyä vaikutelma kookkaasta alaleuasta.
Apertpotilaat hyötyvät kraniofakiaalikirurgiasta.
Otosclerosis
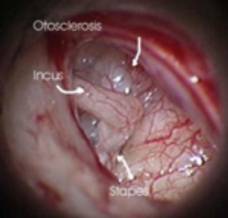
Otoskleroosi ei ole oikeastaan syndrooma. Tahdoin sen kuitenkin ottaa mukaan tähän kirjoitelmaan koska tällä korvakirurgeja kiinnostavalla sairaudella on yhteys geeneihin.
Otoskleroosi voi johtaa konduktiiviseen ja sensorineuraaliseen kuulonlaskuun. Ongelmana ovat endokondraalisen temporaaliluun pesäkkeet, jotka alkuun ovat spongioottisia mutta vähitellen muuttuvat kovaksi skleroottiseksi luuksi. Pesäkkeet aiheuttavat herkästi jalustimen fiksaation soikeassa ikkunassa mutta voivat myös vaikuttaa pyöreään ikkunaan. Otoskleroosipesäkkeitä on löydetty myös kokleasta ja niiden vaikutus sisäkorvaan on jonkin verran kiistanalainen.
Väestöstä 0,5 %:n arvioidaan sairastavan otoskleroosia ja post mortem tutkimuksissa muutoksia on löydetty noin 10 %:lta väestöstä. Tauti on yleisin valkoisella rodulla. Sairauden ajatellaan olevan perinnöllinen mutta sen penetranssi ja ilmeneminen vaihtelevat niin, että perinnöllisyyden tarkkaa luonnetta on vaikea arvioida. Tuhkarokkoviruksella oletetaan olevan yhteyttä taudin ilmenemiseen. Naisilla se on vain hiukan yleisempi kuin miehillä. Taudin arvioidaan periytyvän autosomaalisti dominantisti.
Osteogenesis imperfectalla eli hauraiden luiden oireyhtymällä on yhteys myös otoskleroosiin. Potilailla on autosomaalisti dominantisti periytyvä luusairaus. Heillä on tyyppi I kollageenin vajaus. Kollageenin glysiini korvautuu isommilla aminohappomolekyyleillä. Tämä voi johtaa kollageenin hydrolysoimiseen ja luuston haurastumiseen kun luuston hydroksiapatiittikiteiden järjestyminen häiriintyy. Osteogenesis imperfektasta tunnetaan useita alatyyppejä ja joissakin niistä on helposti ulospäin näkyvänä tuntomerkkinä sinertävät sklerat. Potilaiden hoitoon käytetään myös bisfosfanaattivalmisteita. Näiden merkitystä otoskleroosin hoidossa tutkitaan.
Kartagener
Oirekuvan kuvasi Manes Kartagener vuonna 1933. Aikaisemminkin se oli kuvattu (Siewert 1904). Kartagenerin taudin piirteinä ovat: situs inversus, krooninen sivuontelotulehdus ja bronkkiektasiatauti. Sisäelinten asema voi myös olla poikkeava , situs ambliguus.
Sairauden yleisyys on ilmeisesti 1 / 32 000. Periytyminen on autosomaali resessiivinen.
Sen olennaisena piirteenä on primaari värekarvojen dyskinesia (PCD). Ongelman taustalla on suurella osalla potilaista mutaatio dyneiiniä koodaavissa geeneissä DNAI1 tai DNAH5. Huonosti toimivat siliat johtavat liman huonontuneeseen puhdistumiseen hengitysteistä ja sitä kautta toistuviin tulehduksiin. Oirekuvan liittyy usein myös infertiliteetti koska naisilla munasolun kuljetus Fallopian putkessa häiriintyy ja miehellä häiriintyy siittiöiden liikkuvuus. Noin puolella PCD-potilaista esiintyy situs inversus. Oireilu alkaa jo lapsuudessa. Välikorvantulehdus ja sekretooriotiitti kuuluu oirekuvaan tuba Eustachiin toimintahäiriön vuoksi. Nenäpolyyppeja on 30 %:lla potilaista ja TT-kuvaus tuo esiin varjostuneet sivuontelot ja vahvat limakalvovallit. Alahengitysteissä esiintyy toistuvia pneumonioita, kroonista keuhkoputkentulehdusta ja bronkkiektasioita. Sormien kynnet voivat olla kellolasimaiset.
Kuusankoskella 12. 12. 2009
Hannu Tapiovaara
Takaisin Korvalääkärin kotisivujen sisällysluetteloon
